Dissociation Path Construction
Code
Introduction
Important elements that contribute to protein ligand dissociation free energy are intra and intermolecular interactions, conformational rearrengments and concentric motions. Coarse reaction coordinates are incapable of capturing these effects. On the other hand, using all atom positions is computationally expansive and hard to visualize. Thus we employ a method that uses selected atom positions compressed with PCA to preserve variance. Using this reduced dimension we build dissociation path, which is essential initial step. In a reduced space every frame of MD trajectory (Figure 1) is represented as a point (Figure 2).
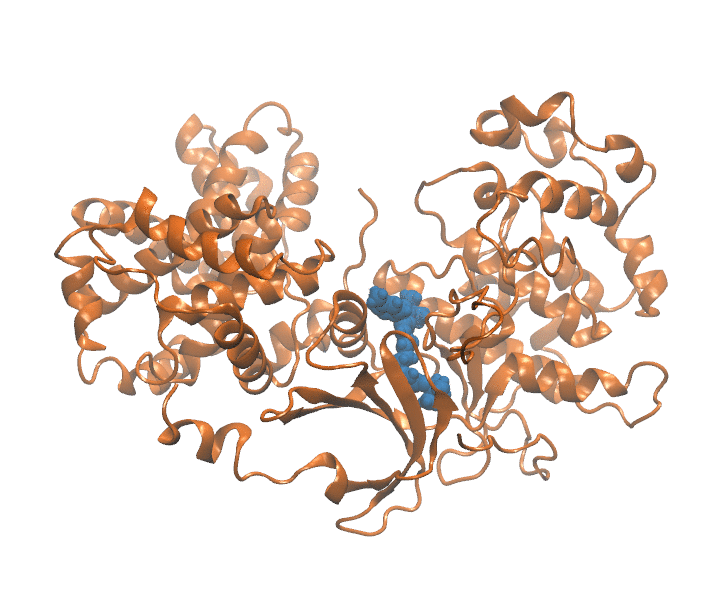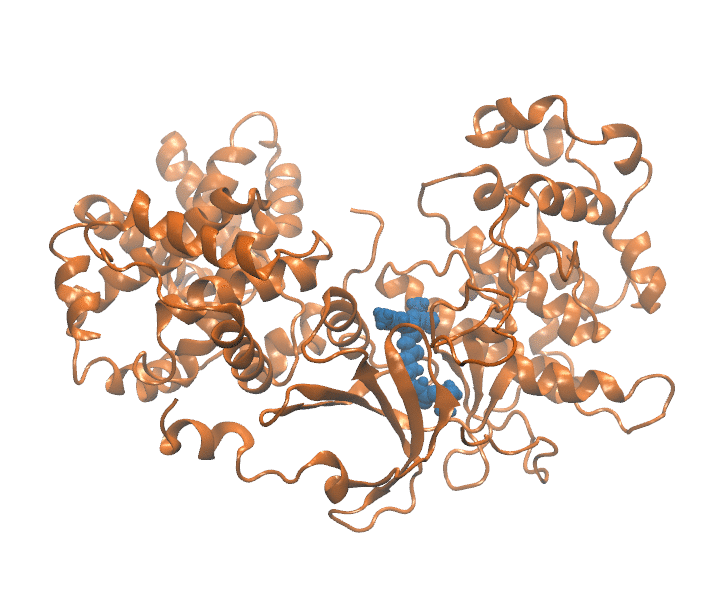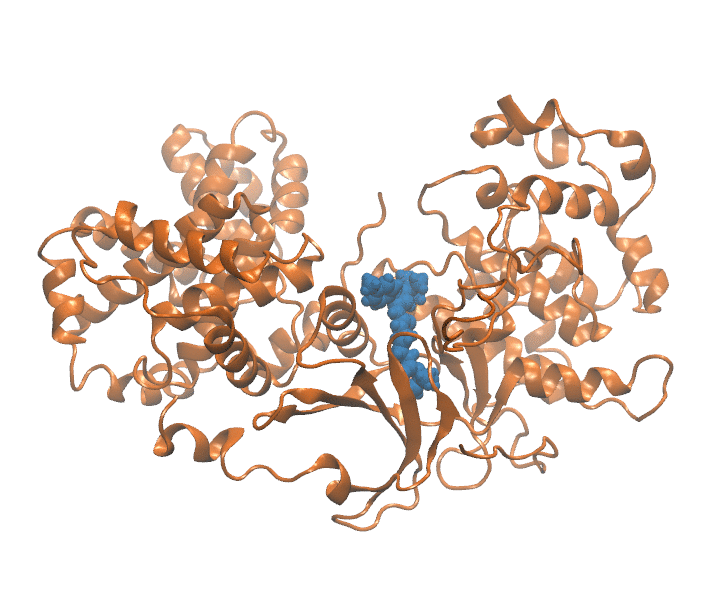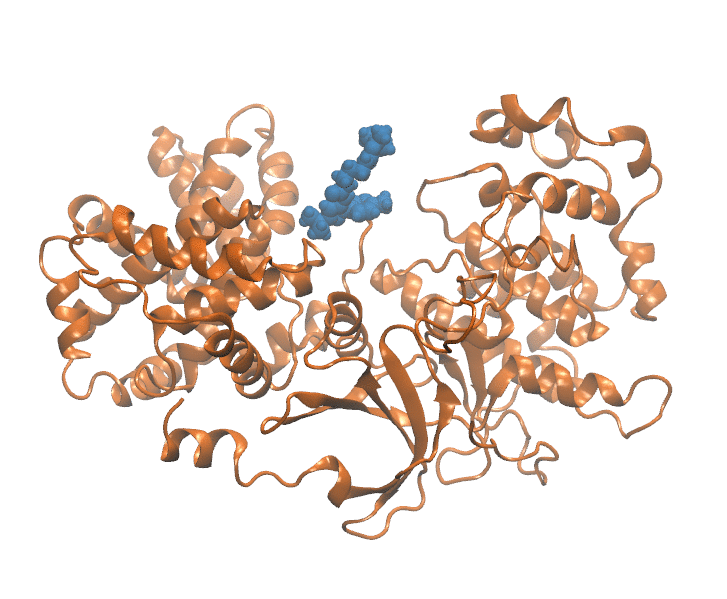
Figure 1: Unbinding transiton visualization for CDK8-PL1 complex.
MD Trajectories to Principal Components
Molecular dynamics (MD) unbinding trajectory contains information about how all atoms of the protein ligand complex transition from bound to unbound state. This information can be used to get an approximate transition path in a PCA space. Here we select backbone $C_{\alpha}$ atom positions of protein and all heavy atoms (non-hydrogen) of ligand to use as our collective variables. 3N-6 number of variables are linearly transformed into PCA space. We keep only two/three components preserving maximum variance and proceed to building dissociation path in a reduced PCA space. (Figure 2).
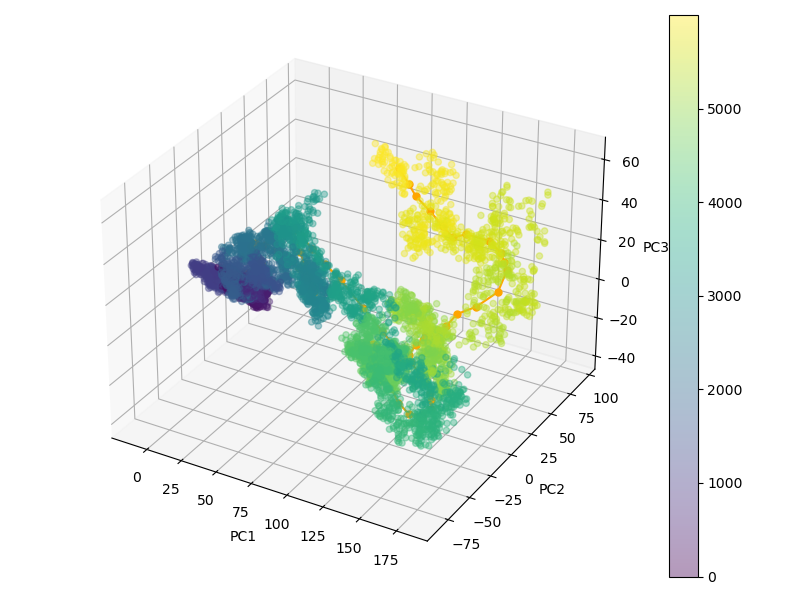
Figure 2: Dissociation trajectories projected into PCA space illustrated, where from dark blue to bright yellow transformation assumes unbinding.
Smooth Unbinding Path
Unbinding path is constructed assuming trajectory indecies (time frame) correspond to distinct protein-ligand conformation, and represent continuous dissociation. In this, we use moving average across window of frames, or points in PCA space to get mean path. Parameters are window size and stride. Window size is number of points considered for taking the average, and stride dictates number of steps to skip between consecutive averages. We set $stride = w_{size}/5$ as default value, but it can be changed. Larger the window size smoother dissociation path we get.
Next we apply another algorithm to smooth the unbinding path. In this, we visit every point and check bond angle it makes with neirest neighbour points in sequence. We systematically remove points if the angle is less than the threshold $\cos(\alpha) = - 0.1$. In order to set equal distance between milestones, we apply interpolation after sharp corners are discarded. (Figure 3) This step is essential preparation for milestone construction that is discussed in the next chapter.
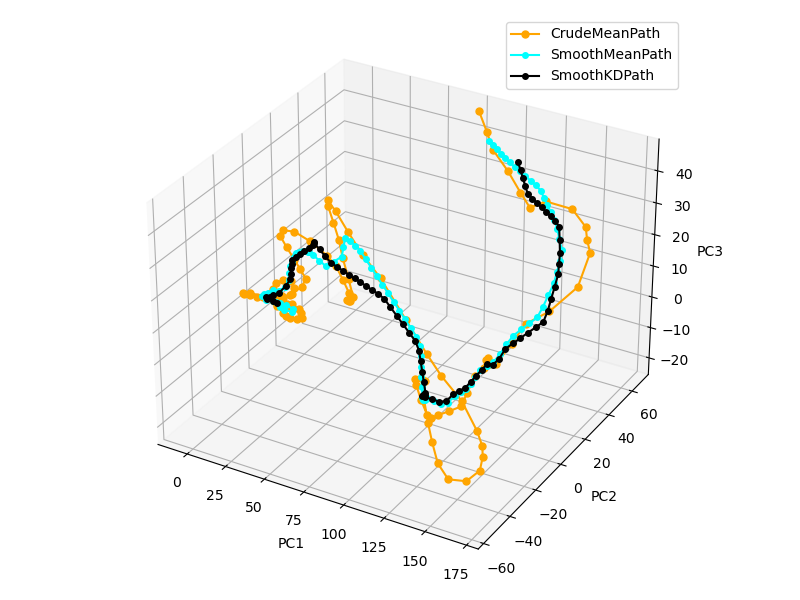
Figure 3. Smooth mean path constructed for CDK8-PL1 complex dissociation trajectory in PCA space with $w_{size}=550$ and $stride=110$. Crude mean path with twice smaller $w_{size}$ and $stride$ is illustrated in without smoothing steps.
KDTree Based Average Path
Due to complex nature of interactions molecular dynamics dissociation trajectory often contains backward motion, where frame indecies inversly correlate with dissociation reaction coordinate. Thus, frame indecies fail to represent dissociation as continuous variable. We provide a robust approach that constructes unbinding path based on local distance average using KDTree algorithm, which facilitates fast neighbour search. In this we use previously constructed mean path as reference and re-evaluate every point by calculating k-neirest neighbour average on original data points. as shown in Figure 3, black color. We restrict neighbourhood size to disc radius. This approach is also helpful when there are multiple transition pathways.